
Tuberous sclerosis
Tuberous sclerosis is a genetically determined disease from the group of phakomatoses, with a wide range of clinical manifestations, accompanied by the development of multiple benign tumors (hamartomas) in various organs, including the brain, eyes, skin, heart, kidneys, liver, lungs, gastrointestinal tract, endocrine and skeletal systems.
Tumors, gradually progressing and increasing in size, disrupt the functions of organs, sometimes leading to fatal consequences. It is a chronic progressive life-threatening disease that leads to reduced life expectancy and disability for patients.
Etiology. Tuberous sclerosis is an autosomal dominant genetically heterogeneous disease with a high incidence of new (spontaneous) mutations, which are found in 2/3 of patients. There are tuberous sclerosis type 1 caused by a mutation in the TSC1 gene and tuberous sclerosis type 2 caused by a mutation in the TSC2 gene. Approximately 10 to 30% of cases of tuberous sclerosis are caused by mutations in the TSC1 gene (OMIM 605284) (tuberous sclerosis type 1, OMIM # 191100), located on chromosome 9 in region 9q34, which codes for the protein gamartin. The remaining cases of the disease are caused by mutations in the TSC2 gene (OMIM 191092) (tuberous sclerosis type 2 - OMIM # 613254), localized on chromosome 16 in the 16p13 region and encoding the tuberin protein. The TS genes are characterized by high penetrance (up to 100%) and variable expressivity (in relatives with the same familial dominant mutation, the severity of the disease may differ).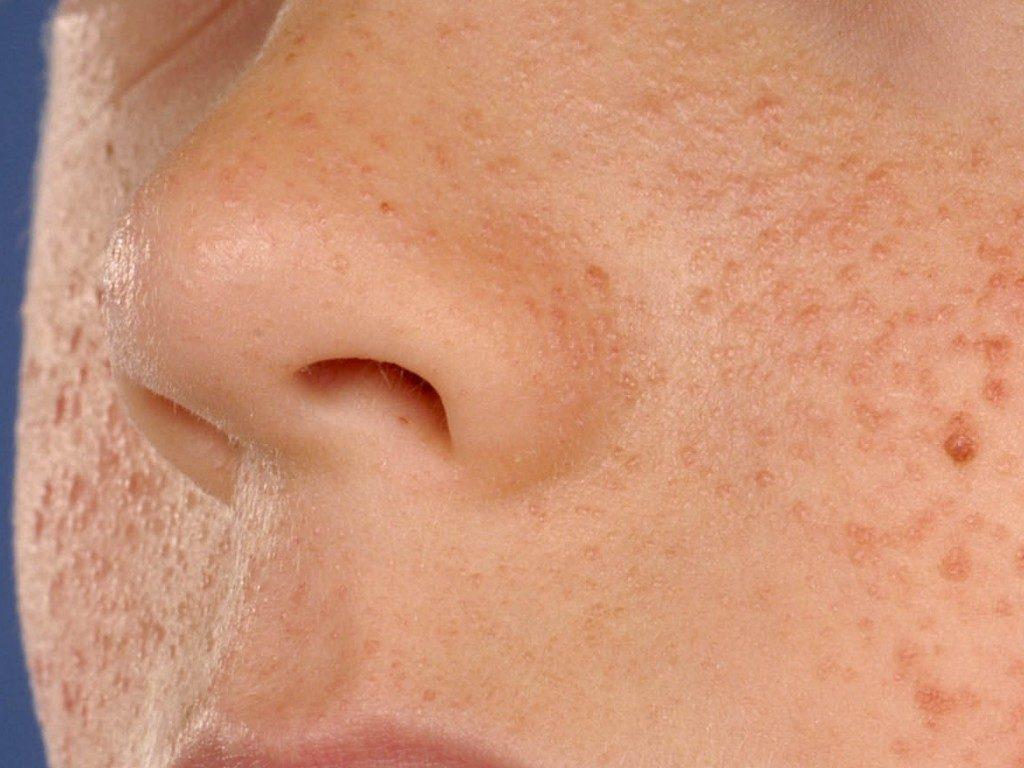
Pathogenesis. The TSC1 and TSC2 genes are normally natural tumor suppressor genes. The protein products of the TSC1 and TSC2 genes, hamartin and tuberin, form a heterodimer capable of inhibiting the signaling cascade mediated by the mammalian Target of Rapamycin Complex 1 (mTORС1) complex. The mechanism of TS pathogenesis consists in mutations in the TCS1 and TSC2 genes with the loss of their function and pathological activation of the mTOR kinase associated with mutations. As a result, the PI3K / Akt / mTOR signaling pathway is activated. This cascade is a key regulator of cell growth and proliferation. It is activated in response to nutrient and growth factors, regulating a number of cellular functions such as translation, transcription, and autophagy. Hyperactivation of the mTORС1 cascade, leading to increased cell proliferation, is considered an important link in malignant transformation (Fig. 1). In cells with mutations in TSC1 and TSC2, this signaling pathway is constantly “on”. This signal transduction pathway is a key link in the pathogenesis of TS. One of the mutations TSC1 and TSC2 is found in all cells of the body. In the progenitor cell of the tumor clone, the second allele, unaffected by the hereditary mutation, is inactivated.
Symptoms_2.jpg) • angiofibromas of the face (at least three) sludge and fibrous plaques on the head;
• angiofibromas of the face (at least three) sludge and fibrous plaques on the head;
• hypopigmented spots (not less than three and not less than 5 mm in diameter);
• non-traumatic periungual fibromas (at least two);
• a section of "pebbled skin";
• multiple retinal hamartomas;
• cortical dysplasias (at least three): cortical tubers and migratory tracts in the white matter • of the brain;
• subependymal nodes (at least two);
• subependymal giant cell astrocytoma;
• multiple or single rhabdomyomas of the heart;
• lymphangioleiomyomatosis of the lungs;
• multiple renal angiomyolipomas (at least two).
Diagnosis is based on a combination of clinical symptoms and symptoms detected during additional laboratory examination. The phenotype of a patient with TS depends on the number, location, and size of hamartomas. The age of the patient also plays an important role as different symptoms of the disease appear at different age periods. The large number of clinical signs of TS, variability of the phenotype, and the fact that the manifestation of signs depends on the patient's age complicates the diagnosis of the disease.
Confirmed pathogenic mutation TSC1 or TSC2 is the main criterion sufficient for the diagnosis of tuberous sclerosis.